Navigating CE Marking and Global Compliance for DHTs in Clinical Trials
Introduction

CE marking is the entry ticket — not the compliance destination. Understanding the full regulatory journey for DHTs across EU MDR, FDA, MHRA, and beyond.
Introduction: The CE Marking Misconception
You’ve got the CE mark. Congratulations — you’ve passed the velvet rope into one of the world’s most demanding regulatory clubs. But here’s the uncomfortable truth that catches sponsors mid-stride: that CE mark on your wearable, your eCOA app, or your connected spirometer does not mean you’re compliant everywhere. Not in the US. Not in Japan. Not even, post-Brexit, in the UK without some additional paperwork [1,2].
For sponsors deploying Digital Health Technologies (DHTs) in multinational clinical trials, CE marking is less a finish line and more a starting block. This article compares the regulatory frameworks that govern DHTs across four major jurisdictions — the EU, the US, the UK, and the rest of the world — and provides a practical roadmap for navigating them without losing your study timeline or your regulatory composure.
Because in this business, regulatory surprise is never the good kind.
1. CE Marking: What It Actually Proves
CE marking under the EU Medical Device Regulation (MDR 2017/745) certifies that a device conforms with General Safety and Performance Requirements (GSPRs) [1]. For DHTs — especially software classified under MDR Rule 11 — this means the device has been assessed for safety, performance, and clinical benefit, either through self-certification (Class I) or Notified Body review (Class IIa and above).
In clinical trials, CE marking fulfils two functions:
Demonstrating baseline safety and performance. Regulators, ethics committees, and investigators gain confidence that the device has been independently evaluated [1].
Supporting endpoint acceptance. Device-derived data is more likely to be accepted in regulatory submissions when the underlying DHT carries recognised conformity certification [3].
But CE marking has a jurisdictional ceiling. It tells regulators what the device is and does within the EU — not whether it meets the unique requirements of the FDA, MHRA, Health Canada, NMPA, or PMDA.
— Key Takeaway
CE marking is proof of EU conformity. It is not a global passport. For every jurisdiction beyond the EEA, sponsors must build a parallel evidence trail.
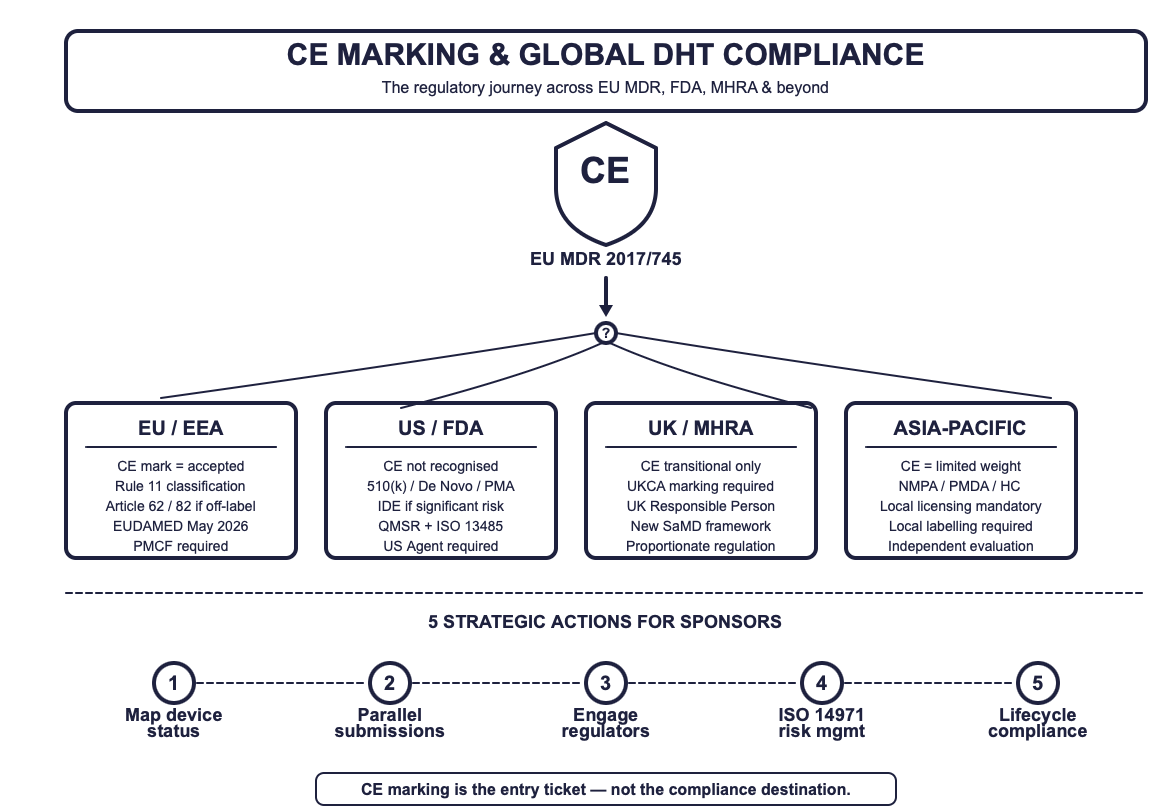
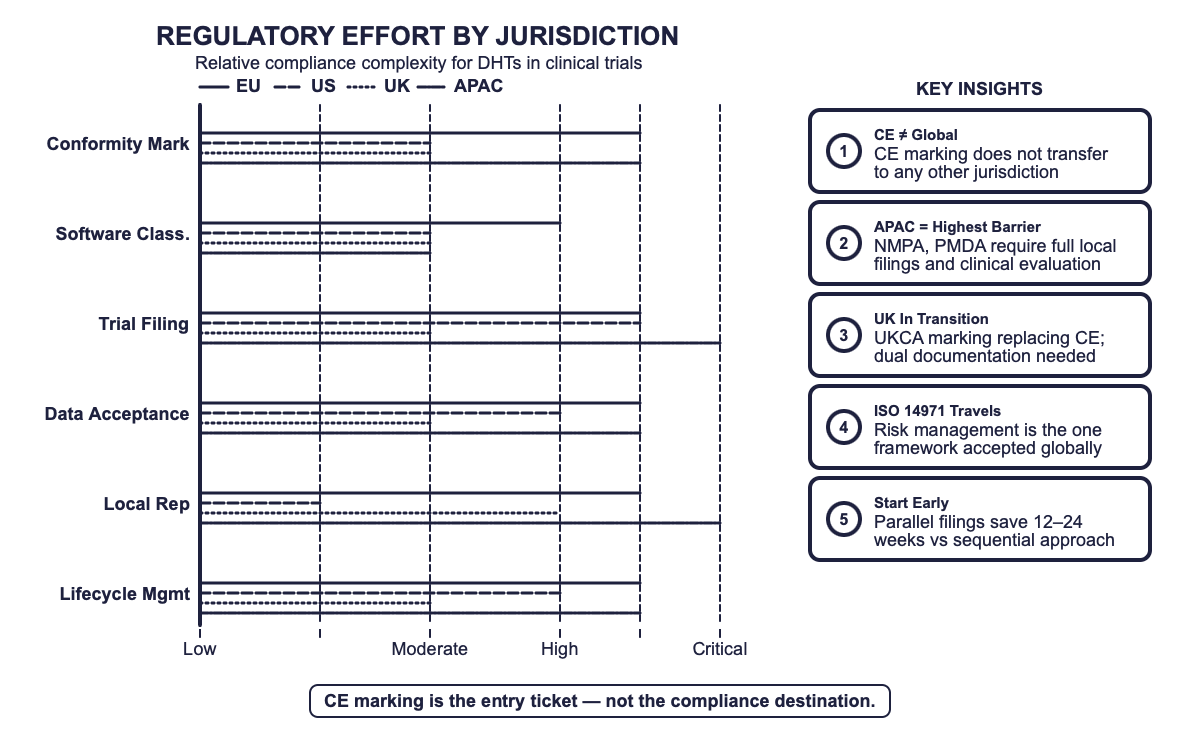
2. The Global Comparison: How Four Jurisdictions Treat DHTs
The following table compares how CE marking, FDA clearance, UKCA marking, and other major regulatory frameworks apply to DHTs used in clinical trials. Use it as a decision matrix when planning multi-country deployments.
| Aspect | EU (MDR) | US (FDA) | UK (MHRA) | Canada / Asia-Pacific |
|---|---|---|---|---|
| Conformity Mark | CE Mark (MDR 2017/745) | FDA Clearance (510(k), De Novo, PMA) | UKCA Mark (transitional CE accepted) | Jurisdiction-specific (Health Canada, NMPA, PMDA) |
| Software Rule | Rule 11 — often Class IIa+ | Risk-based (IMDRF-aligned) | Aligned with MDR; transitioning | Independent classification systems |
| Trial Requirement | CE mark or Article 62/82 filing | IDE if significant risk; exempt if cleared | UKCA/CE + MHRA registration | Local licensing mandatory [4,5,6] |
| Data Acceptance | CE data accepted; PMCF required | Own clinical evaluation; CE data may supplement | CE data accepted during transition | Local clinical evaluation often required |
| Responsible Person | EU Authorised Representative (Art. 11) | US Agent for foreign manufacturers | UK Responsible Person required | Local representatives mandatory |
| Key Risk | Article 62 triggered by off-label use | IDE delays for non-cleared devices | UKCA transition timeline uncertainty | Language, labelling, local trials [5,6] |
3. The Myth of Global Equivalence
FDA: “Show Me Your Own Papers”
The FDA does not recognise CE marking as evidence of compliance. A device cleared in Europe still requires US-specific clearance — 510(k), De Novo, or PMA — before it can be legally marketed or, in many cases, used in US trial arms. If the DHT is investigational or poses significant risk, an Investigational Device Exemption (IDE) under 21 CFR Part 812 is required [2]. CE documentation can support a submission, but it cannot replace one.
MHRA: “We’re Transitioning, Please Hold”
Post-Brexit, the MHRA continues to accept CE marking during a transitional period but is phasing in the UKCA mark. DHTs used in UK trials must be registered with the MHRA, and overseas manufacturers must appoint a UK Responsible Person [3]. The regulatory framework is in flux — with a new UK-specific SaMD classification expected to take shape in 2025–2026 — meaning sponsors need dual documentation strategies.
Canada, China, Japan: “We Have Our Own System, Thanks”
Health Canada requires formal device licensing under SOR/98-282; CE-marked evidence may inform dossiers but does not replace local authorisation [4]. China’s NMPA and Japan’s PMDA operate independent review systems where CE marking carries limited weight. Local clinical evaluation, language-specific labelling, and in-country registration are the norm, not the exception [5,6].
4. When CE Marking Isn’t Enough: Three Real-World Scenarios
Scenario A: Wearable in Oncology — EU vs. US
A CE-marked wearable for remote patient monitoring was accepted in Germany under MDR documentation. The US arm of the same trial required an IDE submission because the device lacked FDA clearance. Result: six-month delay on the US side while the IDE was processed [2].
Scenario B: eCOA Platform — Multinational Deployment
An electronic clinical outcome assessment platform, CE-marked under MDR, faced additional registration requirements in Canada and Japan. Sponsors prepared parallel submissions to avoid study delays, absorbing an estimated 12–16 weeks of additional regulatory lead time [4,6].
Scenario C: Post-Brexit Spirometry — EU vs. UK
CE-marked digital spirometers used in a respiratory trial were accepted across EU sites but required UKCA transition planning in the UK. Sponsors developed dual documentation strategies, effectively managing two parallel conformity pathways for a single device [3].
5. Decision Framework: When to Do What
Not every jurisdiction requires the same depth of effort. The following decision criteria help sponsors prioritise their regulatory strategy:
| Question | If Yes | If No |
|---|---|---|
| Is the device CE-marked for the intended use? | Proceed in EU/EEA; confirm validity for trial context | Consider Article 62 or Article 82 filing under MDR |
| Does the trial include US sites? | Verify FDA clearance status; prepare IDE if needed | No FDA action required |
| Does the trial include UK sites? | Confirm UKCA/CE transitional acceptance; appoint UK RP | No MHRA action required |
| Is the device used outside its declared intended purpose? | Reclassification risk: seek regulatory advice early | Continue with standard conformity pathway |
| Are there sites in Canada, China, or Japan? | Initiate local licensing in parallel; budget 12–24 weeks | No additional filings needed |
6. Strategic Considerations for Sponsors
Map device status by jurisdiction before study start-up. Do not assume CE marking travels. Confirm clearance, registration, and marking requirements for every country in the trial footprint [1,2,3].
Prepare for dual (or triple) submissions. CE documentation can support dossiers but should never be the sole evidence base. Parallel submissions in Canada, Japan, and China should start early [4,5,6].
Engage regulators proactively. Use FDA Q-submissions, EMA scientific advice, MHRA Innovation Office consultations, and PMDA pre-submission meetings. Early engagement prevents late-stage surprises [3].
Embed ISO 14971 risk management across all jurisdictions. A robust, harmonised risk-benefit file is the single most portable piece of evidence in global submissions [7].
Plan for lifecycle compliance — not just market entry. Software updates, labelling changes, AI/ML model retraining, and supply chain shifts can trigger new obligations in multiple regions simultaneously. Build change control into your DHT governance from day one.
7. What’s Changing in 2025–2027
The regulatory landscape is not standing still. Key developments that sponsors should track:
EU: The European Commission proposed amendments to the MDR and IVDR in December 2025, aiming to simplify clinical investigation procedures, streamline combined studies involving medicinal products and devices, and introduce breakthrough/orphan device designations. EUDAMED’s first four modules become mandatory on 28 May 2026 [8,9].
UK: The MHRA is expected to implement a revised SaMD classification framework in 2025–2026, moving further from EU MDR alignment toward a UK-specific, proportionate regulation model [3].
US: The FDA’s adoption of the Quality Management System Regulation (QMSR) aligning with ISO 13485:2016 is a major harmonisation milestone. Companies implementing ISO 13485 now will benefit in both EU and US markets [10].
Cybersecurity: Under the proposed EU amendments, serious incidents involving actively exploited vulnerabilities will need to be reported to national cybersecurity response teams (CSIRTs) and ENISA through EUDAMED [9].
Conclusion: CE Is the Foundation, Not the Destination
CE marking remains the gold standard of device conformity within the EU. But for sponsors operating across borders, it is a foundation upon which multi-jurisdictional compliance must be built — not assumed.
The sponsors who thrive in this landscape are those who treat regulatory strategy as a clinical operations function — embedded early, resourced properly, and monitored continuously. They map device status before first-patient-in, not after the first audit finding. They engage regulators before submitting, not after a Clinical Hold or a hold on their trial.
CE marking got you through the front door. Now the real work begins.
Disclaimer: This article is intended for informational purposes and does not constitute legal or regulatory advice. Organisations should consult qualified regulatory professionals for jurisdiction-specific guidance.
References
1. European Commission. Regulation (EU) 2017/745 on medical devices (MDR). Brussels: EC; 2017.
2. FDA. Investigational device exemptions (IDE) regulations. 21 CFR Part 812. Silver Spring, MD: FDA; 2023.
3. MHRA. UK medical device regulations and transitional guidance. London: MHRA; 2023.
4. Health Canada. Medical Devices Regulations SOR/98-282. Ottawa: Health Canada; 2023.
5. NMPA. Order 739: Regulations for the supervision and administration of medical devices. Beijing: NMPA; 2021.
6. PMDA. Guidelines on medical device clinical evaluation. Tokyo: PMDA; 2014.
7. ISO 14971:2019. Medical devices — Application of risk management to medical devices. Geneva: ISO; 2019.
8. European Commission. Proposed Regulation amending MDR (EU) 2017/745 and IVDR (EU) 2017/746. Brussels: EC; December 2025.
9. Baker McKenzie. The EU’s 2025 proposal to simplify the Medical and In-Vitro-Diagnostic Devices Regulations (MDR/IVDR). December 2025.
10. FDA. Quality Management System Regulation (QMSR) — Adoption of ISO 13485:2016. Silver Spring, MD: FDA; 2024.