Direct-to-Patient Devices Done Right: Logistics, Traceability & Reuse Without Data Breaks
A playbook for packaging/shipping, returns, sanitization, and chain-of-identity that stands up to audits in decentralized trials.
Introduction

Why This Playbook Exists
Decentralised clinical trials promise speed, reach, and patient-centricity. But every device that leaves a controlled warehouse and arrives on a participant’s doorstep crosses a regulatory fault line. The supply chain is no longer just a supply chain—it is a data chain, and every handover point is a potential data break [1,2].
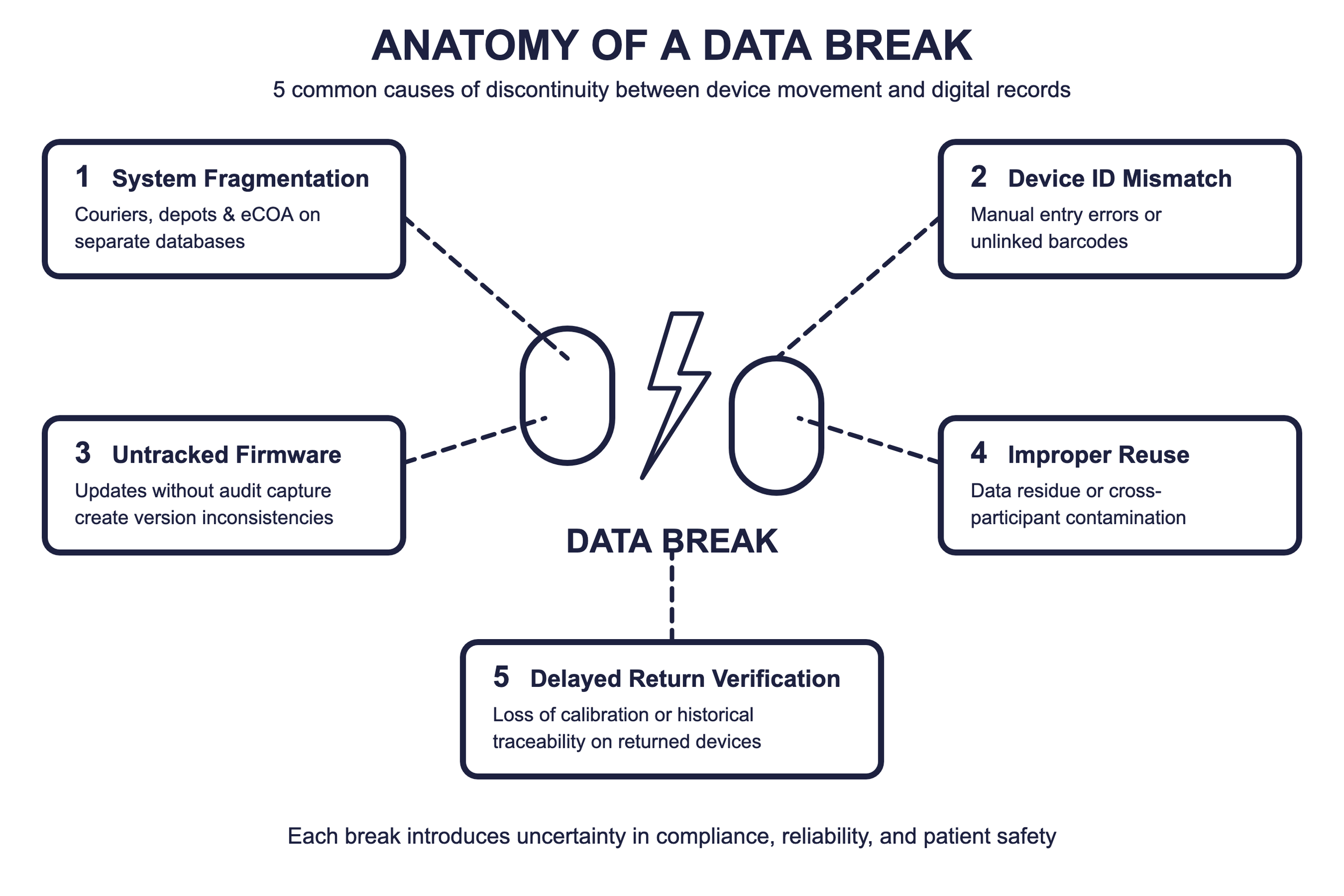
A data break occurs when there is a discontinuity between physical device movement and the digital record that supports it—missing audit trails, mismatched device identifiers, or incomplete datasets that undermine ALCOA+ compliance [3,4]. Regulators including the FDA, EMA, and MHRA now require that the movement of devices and data be seamless, documented, and inspection-ready under Good Clinical Practice [2,5].
This playbook is designed for clinical operations leads, device logistics managers, and quality assurance professionals who need actionable, step-by-step tactics—not theory—to keep their direct-to-patient (DTP) device programmes compliant.
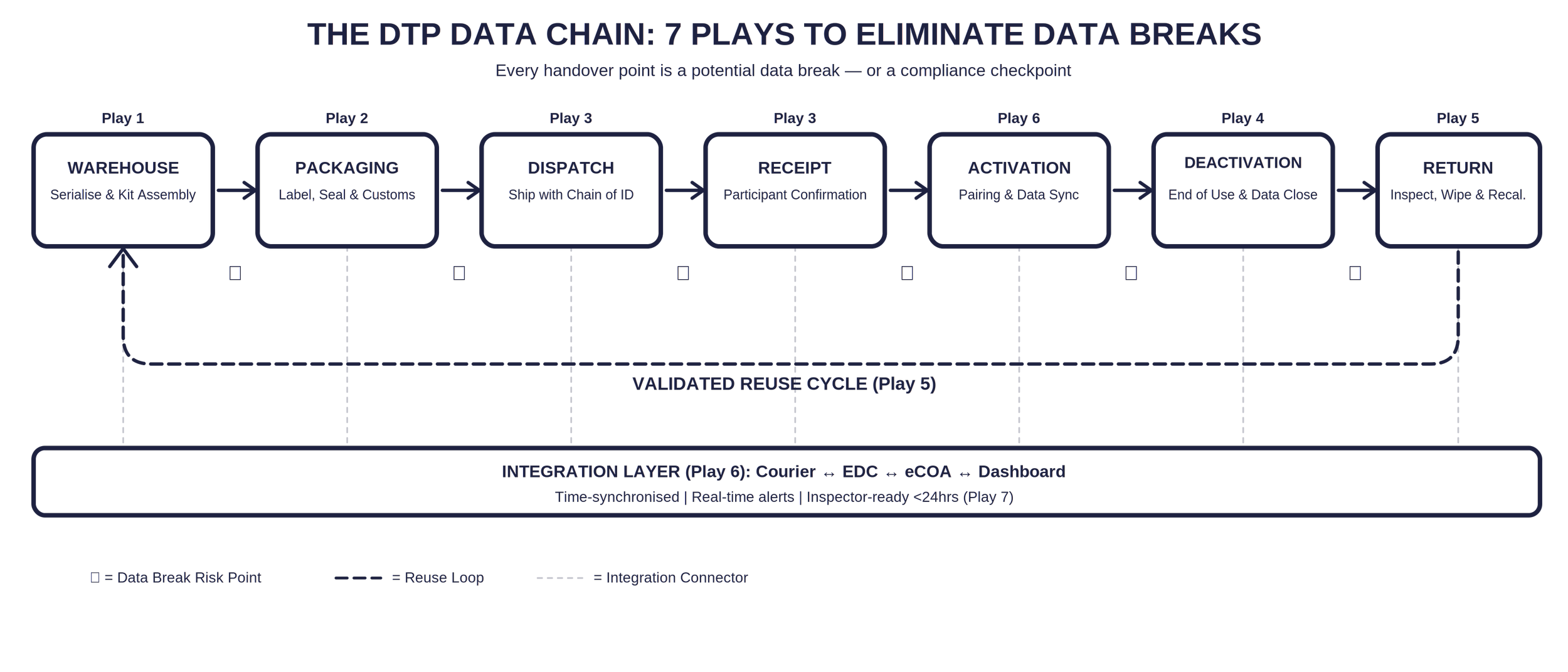
Play 1: Serialise Everything — Before It Leaves the Warehouse
The foundation of traceability is unique identification. Every digital health technology (DHT) shipped in a trial must carry a unique device identifier (UDI) or serial number linked to the participant, trial site, shipment record, and protocol timepoint [6,7].
What to Do
Assign a UDI or serialised QR code to each device at the point of kit assembly.
Integrate barcode or RFID tracking directly with your EDC/eCOA platform so that scanning a device auto-populates the clinical database [7,8].
Ensure identifiers remain visible and scannable across all packaging layers and through every reuse cycle [6].
Test readability of labels under simulated shipping conditions (humidity, abrasion, temperature range).
Field Insight: In a global oncology trial, a wearable sensor programme using serialised QR codes reduced device misallocation errors by 93% [9]. The investment in serialisation paid for itself in the first quarter.
Play 2: Package for Compliance, Not Just Protection
Packaging in DTP logistics is not merely protective—it is evidentiary. The exterior of every kit is the first thing a customs officer, site coordinator, or patient sees. If the labelling is wrong, incomplete, or in the wrong language, the device may never reach the participant—or worse, it arrives and generates non-compliant data [10,11].
Labelling Checklist
Regional regulatory markings (CE, UKCA, FDA clearance identifiers) visible on outer packaging [6,11].
Protocol number and study identifier on the shipping label and internal device card.
Instructions for Use (IFU) in the participant’s native language, aligned with EU MDR Annex I and GSPR requirements [6,11].
Tamper-evident seals that provide visual confirmation of integrity on receipt.
Temperature or shock indicators for sensitive devices (cold-chain or vibration-sensitive DHTs) [12].
Return prepaid shipping materials and instructions inside every outbound kit.
Field Insight: Direct-to-Patient always includes Direct-to-Customs, whether you planned for it or not. Build customs documentation into your kit assembly SOP—not as an afterthought.
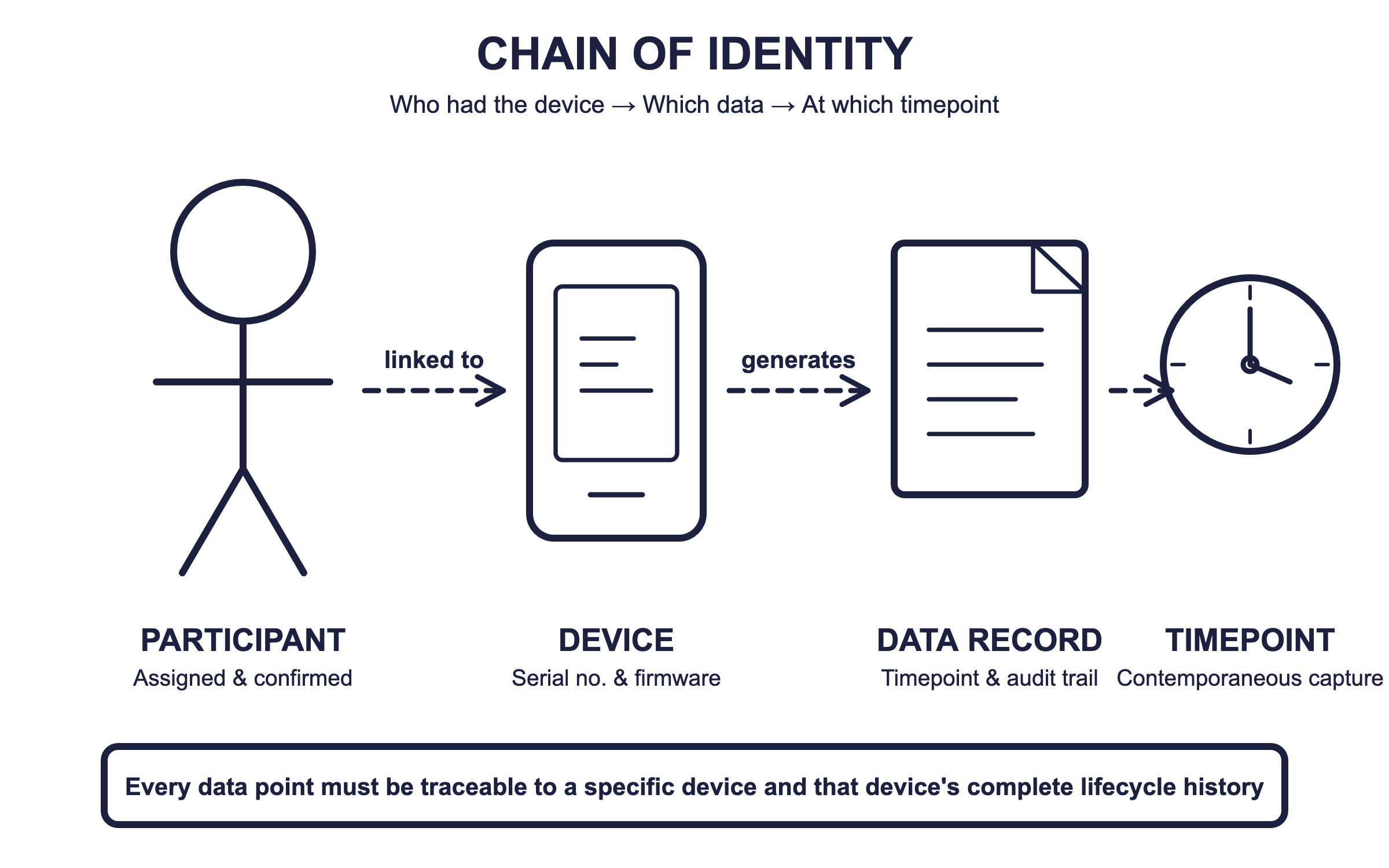
Play 3: Build an Unbroken Chain of Identity
Chain of identity goes beyond chain of custody. While custody tracks who had the device, chain of identity tracks which specific device was in whose hands, generating which data, at which timepoint [2,7]. Every data point must be traceable to a physical device and that device’s
The Five Handover Points You Must Capture
| Handover | What to Record | Regulatory Anchor |
|---|---|---|
| 1. Dispatch | Serial no., firmware version, calibration date, courier ID, timestamp | FDA 21 CFR 820; ISO 13485 [6,7] |
| 2. Receipt | Participant confirmation (e-signature or photo proof), condition check, tamper seal intact | ICH E6(R3); EU MDR Art. 10–14 [2,5] |
| 3. Activation | Device pairing event, time-sync verification, first data capture confirmation | 21 CFR Part 11; EMA e-data guideline [13,14] |
| 4. Deactivation | Last data capture timestamp, device status, reason for deactivation | ALCOA+ principles; ICH E6(R3) [5,15] |
| 5. Return | Return shipment tracking, condition assessment, data wipe confirmation, recalibration status | ISO 14971; ISO 13485 [7,16] |
Play 4: Design Reverse Logistics as a Data Process, Not a Shipping Process
Most logistics plans focus on getting devices out. Far fewer address getting them back—and that is where data chains break most often. Returns generate more questions than shipments, and every unreturned device is a potential audit finding [4,16].
Reverse Logistics Workflow
Pre-stage return kits: Include prepaid return packaging, return instructions, and a data wipe confirmation card inside the original shipment.
Automate return triggers: Set EDC or eCOA system alerts when a participant reaches end-of-treatment or early discontinuation, prompting the return logistics sequence [8].
Track return in real time: Use the same courier-to-EDC integration used for outbound shipments. Cloud-based dashboards should flag unreturned devices after a defined grace period [8,9].
Inspect and document upon receipt: Record physical condition, battery status, firmware version, and any visible damage. Archive photographs for the quality dossier [7,16].
Validated data wipe: Execute secure data deletion using a validated script. Record the wipe event with timestamp and operator ID. This is not optional—it is a regulatory expectation under GDPR, Part 11, and ISO 27001 [13].
Play 5: Validate Reuse — Don’t Just Assume It Works
Device reuse is both a sustainability win and a cost optimiser—but only when it is treated as a regulated process. Reusing devices reduces hardware costs (one study reported a 45% reduction [18]), yet sponsors must prove that reuse does not compromise data quality or participant safety [7,16].
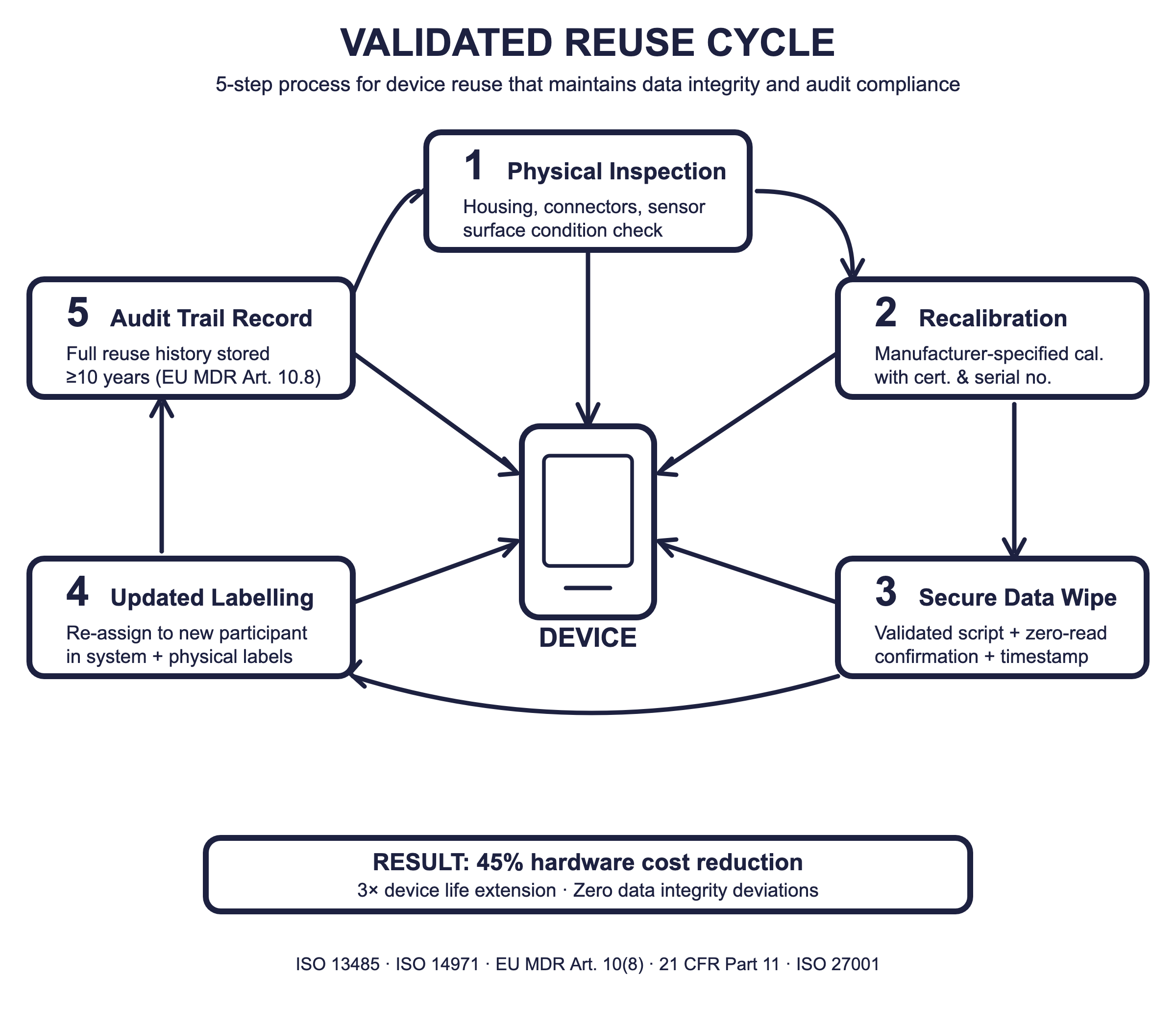
Reuse Validation Cycle
Physical inspection: Check housing integrity, connector wear, sensor surface condition.
Recalibration: Perform manufacturer-specified recalibration. Document calibration certificate with device serial number and date [7].
Secure data deletion: Execute validated wipe; verify with test read confirming zero residual data [13].
Updated labelling: Re-assign device to new participant in the system. Update physical and electronic labels [6,11].
Audit trail: Record the complete reuse history—including which participants previously used the device, dates, and all reconditioning steps—for a minimum of 10 years per EU MDR Article 10(8) [6].
Field Insight: In a metabolic monitoring study, validated device reuse extended device life threefold with zero data integrity deviations [18]. Green trials can be clean trials—if the SOPs are rigorous.
Play 6: Integrate Logistics Tracking with Clinical Data Capture
The most common cause of data breaks is system fragmentation: couriers, depots, and eCOA platforms operating on separate databases with no shared source of truth [3,4]. The solution is integration—not just between systems, but between the logistics mindset and the clinical mindset.
Integration Architecture
API-based connections between courier platforms, EDC, and eCOA systems that auto-confirm delivery, activation, and first data capture [8,9].
Cloud-based dashboards providing real-time visibility of device location, status, and performance across all trial sites [8].
Automated alerts for delayed shipments, unreturned devices, inactive participants, and firmware version mismatches [4,9].
Time-synchronised servers maintaining accurate chronological sequencing across all systems, preserving ALCOA+ compliance [13,15].
Field Insight: During COVID-19, sponsors rapidly scaled DTP logistics but faced data inconsistencies because courier and EDC systems were not communicating. API-based dashboard integration later restored end-to-end visibility [19]. Build the integration before you need it.
Play 7: Prepare for the Inspector’s Questions Now
Inspectors do not audit logistics plans. They audit evidence. If you cannot produce a complete device lifecycle narrative—from warehouse shelf to participant’s wrist to validated return—within 24 hours of a request, you have a finding [2,5].
The Questions Your Documentation Must Answer
Can you prove each device was shipped, labelled, and handled in compliance with local laws and GDP standards? [6,10]
What systems track device inventory, usage history, and chain of custody—especially when reusing devices across participants or studies? [7,8]
Have you experienced customs delays due to missing documentation, importer-of-record confusion, or UDI/labelling issues? [10,11]
How are you validating Good Distribution Practice and storage conditions for temperature-sensitive or high-risk devices? [12]
If a regulator asked for proof that your devices were delivered, handled, and reused according to documented SOPs, could you provide that within 24 hours? [2,5]
The Bottom Line: Logistics Done Right Is Data Done Right
In decentralised and hybrid models, every DHT represents a physical embodiment of data. Losing track of a device means losing trust in its data [2,4]. Sponsors who embed end-to-end traceability—from shipment to reuse—create unbroken chains of evidence that satisfy inspectors and build confidence with patients, regulators, and investors.
The seven plays in this playbook are not aspirational. They are operational necessities. The question is not whether your logistics will be inspected, but when—and whether the evidence will be ready.
In the era of connected clinical research, logistics done right is data done right.
References
1. FDA. Investigational device exemptions (IDE) regulations. 21 CFR Part 812. 2023.
2. European Commission. Regulation (EU) 2017/745 on medical devices (MDR). Brussels: EC; 2017.
3. Kellar E, Bornstein S, Celi LA, et al. Optimizing the use of electronic data sources in clinical trials. Ther Innov Regul Sci. 2017;51(4):408–416.
4. Bent B, Goldstein BA, Kibbe WA, Dunn JP. Investigating sources of inaccuracy in wearable optical heart rate sensors. npj Digital Medicine. 2020;3:18. (DOI: 10.1038/s41746-020-0226-6).
5. ICH. E6(R3) Good Clinical Practice draft guideline. International Council for Harmonisation; 2023.
6. ISO 13485:2016. Medical devices – Quality management systems. Geneva: ISO; 2016.
7. ISO 14971:2019. Medical devices – Application of risk management to medical devices. Geneva: ISO; 2019.
8. EMA. Guideline on computerised systems and electronic data in clinical trials. Draft, 2023.
9. Sehrawat O, Noseworthy PA, Siontis KC, et al. Data-driven and technology-enabled trial innovations toward decentralization. Mayo Clin Proc. 2023;98(9):1404–1421.
10. EFPIA. Reflection paper on integrating medical devices into medicinal product clinical trials. Brussels: EFPIA; 2025.
11. MHRA. UK medical device regulations post-Brexit. London: MHRA; 2023.
12. ISO 27001:2022. Information security, cybersecurity, and privacy protection. Geneva: ISO; 2022.
13. FDA. Part 11, electronic records; electronic signatures – Scope and application. Guidance. 2003.
14. EMA. Qualification opinion: stride velocity 95th centile as secondary endpoint in Duchenne muscular dystrophy. EMA; 2019.
15. EFPIA. Reflection paper on integrating medical devices into medicinal product clinical trials. Brussels: EFPIA; 2025.
16. ISO 27001:2022. Information security, cybersecurity, and privacy protection. Geneva: ISO; 2022.
17. Aryal S, Goldsack JC, Izmailova E, et al. Patient centricity in digital measure development. NPJ Digit Med. 2024.