Not in EUDAMED? If You Import the Device, the Obligations Are Already Yours
There is a sentence that has quietly ended more compliance careers than any inspector ever has: “We’ll register once the manufacturer does.”
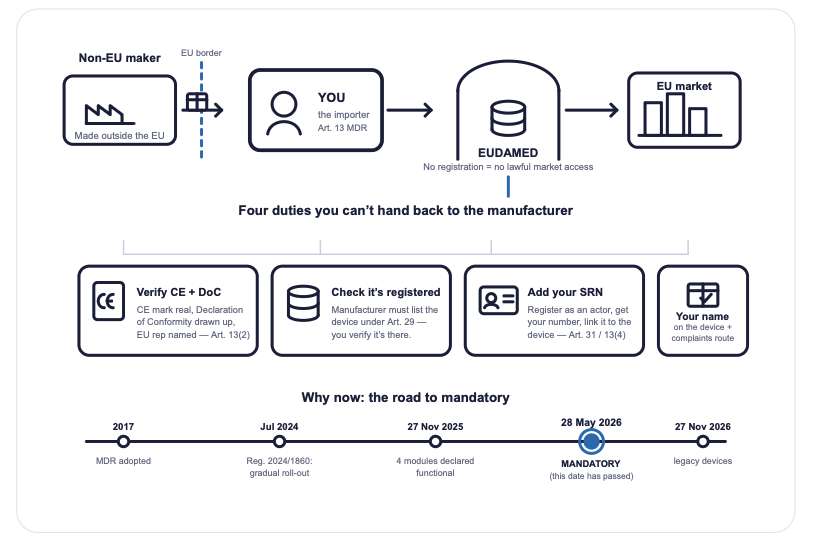
Here is the uncomfortable part. As of 28 May 2026, that sentence is no longer a planning assumption — it is a finding waiting to be written up. On that date, the first four EUDAMED modules became mandatory across the EU. If your organisation brings a non-EU medical device into the Union — a wearable from Shenzhen, a point-of-care reader from a US start-up, a connected sensor that arrived in a clinical-trial kit — then you are an importer under EU MDR Article 13, whether or not anyone in the building has ever used that word.
And the importer does not get to wait for the manufacturer. The importer has independent duties that exist the moment the device crosses the border. This is the playbook for discovering which of those duties are already yours — and closing the gap before someone else points it out.
First, the thing that actually changed
EUDAMED — the European Database on Medical Devices — was written into law back in 2017. For years it lived in a comfortable limbo: technically required, practically optional, perpetually “coming next year.” The original deal was that nothing became mandatory until all six modules were live. They never all went live at once, so the whole thing stayed voluntary, and a generation of importers learned to treat it as someone else’s problem.
Regulation (EU) 2024/1860 broke that pattern. Instead of waiting for the full set, the Commission can now switch modules on one at a time — each becomes mandatory six months after it is independently audited and declared functional. On 27 November 2025, Commission Decision (EU) 2025/2371 declared four modules functional. Six months later, the clock ran out. Here is what is now live:
| Module now mandatory (28 May 2026) | Why an importer cares |
|---|---|
| Actor registration | This is where you register as an economic operator and get your SRN. No SRN, no legitimate place in the chain. |
| UDI / Device registration | You must verify the device is registered here before you place it on the market — and then add your own details. |
| Notified Bodies & Certificates | Where the certificate behind the CE mark actually lives. “It has a CE sticker” is no longer the end of the conversation. |
| Market Surveillance | The module competent authorities use to find you. They will. |
Two modules — Clinical Investigations and Vigilance — are still under development and will switch on under a separate notice. The legacy-device registration deadline lands on 27 November 2026 [3]. So no, this is not “done.” But the parts that catch importers are done.
The role you were assigned without an interview
Nobody sends you a welcome email when you become an importer. There is no onboarding, no badge, no probation period. Under MDR Article 13, the definition is brutally simple: if you are an entity established in the EU and you place a device from outside the Union on the EU market for the first time, you are the importer. The clinical-operations manager who accepted a shipment, the procurement lead who signed the PO, the start-up that “just resold a few units” — all of them have, on at least one occasion, quietly become an economic operator with statutory obligations.
The classic version we see in the field: a device manufactured in China, shipped directly to an EU site, with an EU company’s name on the paperwork. The manufacturer is delighted to let someone else carry the European weight. That someone is you. And the obligations transferred the moment the box arrived — not the moment you got around to reading Article 13.
You don’t opt into being an importer. You discover it. Usually during an audit, which is the most expensive possible moment to learn a definition.
The importer job description (the one you didn’t apply for)
Article 13 is not a vibe — it is a list. Before a device goes on the market, an importer must verify that:
the device carries the CE marking and the EU Declaration of Conformity has been drawn up;
the manufacturer is identified, and an EU authorised representative has been designated where the manufacturer sits outside the Union;
the device is labelled per the Regulation and accompanied by the required instructions for use;
a UDI has been assigned where applicable;
the device is registered in EUDAMED by the manufacturer under Article 29.
And then a set of duties that do not stop at the warehouse door:
Put your name on it. Your registered trade name and a contactable address must appear on the device, its packaging, or an accompanying document — without obscuring the manufacturer’s label.
Protect it in transit. Storage and transport conditions under your control must not compromise compliance.
Keep a complaints register and forward complaints, suspected incidents, and recall information to the manufacturer and authorised representative.
Cooperate with authorities, including providing samples or access on request.
Stop the line. If you have reason to believe a device is not compliant, you must not place it on the market — and you must tell the manufacturer and the authorised representative. If there’s a safety risk, the competent authority too.
Read that last one twice. The importer is not a passive courier. The importer is the gatekeeper — the last EU-established party legally able to say “this does not go on the market.”
Where EUDAMED specifically bites importers
Two articles, one number you can’t fake.
Article 13(4): verify, then add yourself
You must verify that the manufacturer has registered the device in EUDAMED under Article 29 — and then add your own importer details to that registration under Article 31. If the device isn’t there, that’s your problem to surface, because you cannot legitimately place an unregistered device on the market.
Article 31: register as an actor, get your SRN
Importers register as an economic operator in the EUDAMED Actor module and receive a Single Registration Number (SRN). The SRN is not a formality you file and forget. It identifies you across your entire regulatory footprint, and the obligations are continuous: update your data within one week of any change, and confirm its accuracy one year after registration and every two years thereafter. Fail to confirm within six months of those deadlines and a Member State can take corrective measures against you on its own territory.
“The manufacturer registered it” answers exactly one of your obligations. It does not answer the other nine.
Myth vs. fact: “but someone else already imports it”
Here’s the one that trips up procurement teams constantly: there’s already an importer for this device somewhere in the chain — but you buy your units directly from the non-EU manufacturer. Are you off the hook? No. “Placing on the market” is judged per unit, not per device model, so you become an importer for the units you personally bring in.
| The myth | The reality under MDR |
|---|---|
| “Someone else already imports this device, so we’re covered.” |
Importer status attaches to the specific units you bring across the EU border. Another company’s import channel covers its units — not yours. One device model can legitimately have several importers in parallel [5]. |
| “We buy it from an EU company, so we’re only a distributor.” |
Only if the device was already placed on the EU market when you acquired it. If your purchase — direct from the non-EU manufacturer — is what brings the device into the Union, you’re the importer (Art. 13), not a distributor (Art. 14) [5,16]. |
| “The model is already registered in EUDAMED, so there’s nothing to add.” |
Each importer adds its own details and SRN against the device registration under Art. 13(4) / Art. 31. Multiple importers legitimately appear on a single device record [6,16]. |
(The same legal entity can be an importer for one batch and a distributor for another. Role is decided transaction by transaction — which is precisely why organisations lose track of it.)
The test is simple: does the device cross the EU border because of your transaction? If yes, you’re the importer for those units — no matter who else imports the same model.
The 30-minute playbook: what to do this week
You do not need a transformation programme. You need to know whether you have a gap. Run these in order.
Map every non-EU device you touch. Wearables, readers, sensors, accessories, trial kits. If it was manufactured outside the EU and you put it on the EU market first, list it.
Confirm your own SRN exists. Search the EUDAMED Actor module for your organisation. If you have no SRN, you are not yet a recognised economic operator — register today.
For each device, check the manufacturer’s Article 29 registration. Is the device actually in EUDAMED? If not, you have found your blocker before the inspector did.
Add your importer details. Link your SRN to each device record under Article 13(4)/Article 31.
Pull the conformity evidence. CE certificate, EU Declaration of Conformity, the certificate behind it via the Notified Bodies module. “There’s a CE logo on the box” is not evidence.
Check your name is physically on the device flow. Article 13(3) wants a contactable EU address on the device, packaging, or accompanying document.
Stand up a complaints/vigilance route. One inbox, one owner, one path to the manufacturer and AR. Boring. Effective.
What “we’ll do it later” actually costs
Later is a number now. A device that isn’t registered is a device you can’t lawfully place on the market — which means stalled supply, held shipments, and the kind of customs conversation that doesn’t resolve over email. Member States can apply corrective measures and, in several jurisdictions, administrative fines that reach into seven figures for importer non-compliance. And in a clinical-trial context the bill is paid in a currency that hurts more than money: a device tied to a primary endpoint, sitting in a warehouse, while activation slips a quarter.
None of this is exotic. It is the predictable consequence of treating an EU-wide, audited, mandatory database as optional six months after it stopped being optional.
The boring conclusion (the good kind)
EUDAMED didn’t create your obligations. It just made them visible. The duties of an importer have lived in Article 13 since 2017; what changed in May 2026 is that the database where they get checked is now switched on, audited, and enforceable.
So the question isn’t whether you’re in EUDAMED. The question is whether you’ve admitted what role you’re already playing. If you import the device, the obligations are already yours. The only choice left is whether you discover that on your own terms — or in an inspector’s.
If your answer to “who is the importer of record for this device?” is “I’m not sure” — that’s your starting point.