“It’s Just a Phone”
The Assumption That Quietly Pulls You Into Four Regulatory Regimes
Somewhere in your trial, right now, there is a phone. It might be sitting on a kitchen counter in Antwerp, charging beside a bed in Osaka, or tucked into a coat pocket in São Paulo. Someone on the study team called it “just the participant’s phone” and moved on. That sentence — said in good faith, in a feasibility meeting, by a smart person — is one of the most expensive assumptions in modern clinical research.
Here is the uncomfortable truth: the regulators do not care what the device looks like. They care what it does. And the moment that handset is used to collect, transmit, or influence trial data, its physical form stops mattering and its function takes over [1,2]. A phone that captures a six-minute walk test, runs an eCOA, or streams sensor data is no longer consumer electronics. It is an instrument in a regulated experiment — and instruments come with paperwork.
Let’s take the five most common things people believe about study phones, and put each one next to what the regulations actually say.
MYTH 1 “A phone is consumer hardware, so it’s out of regulatory scope.”
FACT Scope is set by intended use and context of use, not by the shelf it was bought from. Under EU MDR 2017/745, software or hardware intended for a medical purpose is a medical device regardless of how ordinary it looks; MDR Rule 11 in particular sweeps decision-supporting software into Class IIa and above [3]. The FDA’s 2023 Digital Health Technologies framework is explicit that a general-purpose platform becomes regulated based on the function it performs in the investigation, and that sponsors must justify fitness-for-purpose for each context of use [1,2]. A consumer smartwatch repurposed for a primary endpoint has triggered Article 62 filings in EU Member States precisely because the intended use expanded [4]. The casing is consumer. The obligations are not.
MYTH 2 “We didn’t buy it as a medical device, so it isn’t one.”
FACT Classification is an outcome of what the device does in your protocol, not a property stamped on the box. The FDA requires context-specific intended-use validation: a device cleared or CE-marked for one purpose must be re-evaluated for each new study context [1]. Once data from a consumer-grade device feeds a regulated endpoint or appears in a submission dossier, the sponsor inherits responsibility for performance, interoperability, and data quality — not the original manufacturer [2]. “We didn’t mean to” is not a regulatory defence. Intended use is judged by how the device is actually deployed.
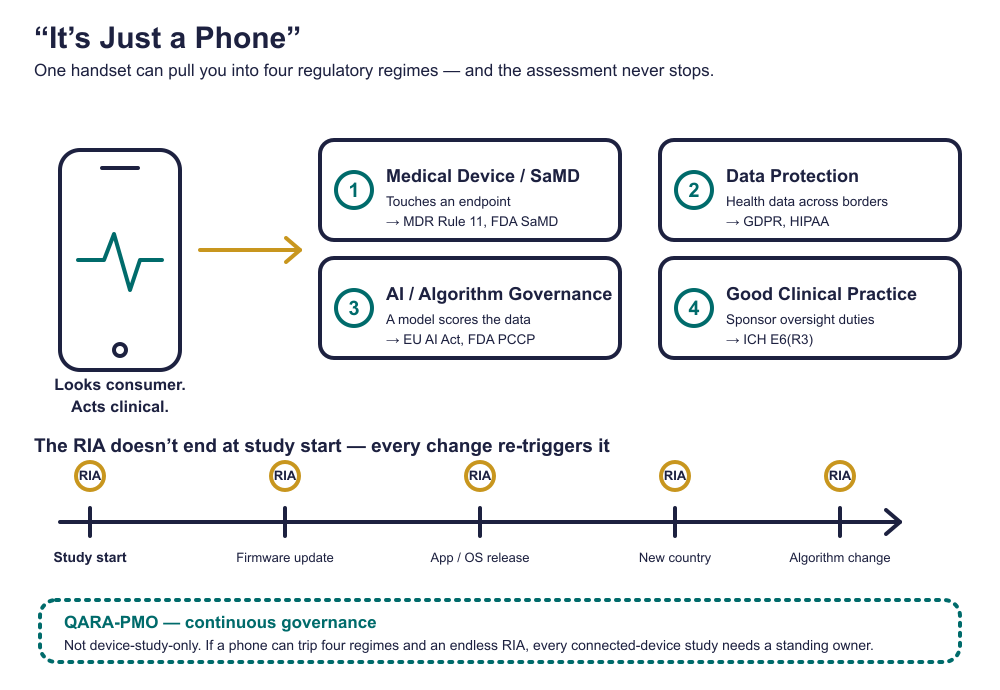
The Four Regulatory Regimes Hiding in One Handset
Here is why “just a phone” is so dangerous: a single device does not pull you into one regulatory conversation. It can pull you into four at once, each with its own authority, its own evidence expectations, and its own inspection. Most teams plan for zero of them.
| Regime | What triggers it | Key references |
|---|---|---|
| 1. Medical device / SaMD |
The phone or its app collects, calculates, or supports an endpoint or safety signal — it becomes (Software as a) Medical Device. |
EU MDR 2017/745, Rule 11; FDA 21 CFR 812/820; IEC 62304 [3,5,6] |
| 2. Data protection & privacy |
Personal and health data are captured, processed, and transmitted across borders. |
GDPR; HIPAA; local laws (APPI, PIPL) [2] |
| 3. AI / algorithm governance |
An app uses ML or an adaptive algorithm to derive or score data. |
EU AI Act; FDA PCCP [2,7] |
| 4. Good Clinical Practice |
The device is embedded in a GCP-governed trial, creating sponsor oversight duties. |
ICH E6(R3); FDA DHT Framework [1,8] |
One handset. Four authorities. Each can stop your trial independently.
MYTH 3 “One Regulatory Impact Assessment at study start covers us.”
FACT This is the assumption that ages worst. A Regulatory Impact Assessment (RIA) is not a gate you pass once — it is a living obligation. Firmware updates, app releases, operating-system upgrades, algorithm changes, added countries, and device substitutions are all regulatory events that can silently break endpoint comparability mid-study [5,7]. In modern trials the software is the instrument, and an instrument that changes after baseline changes your evidence [6]. EU MDR Article 22 and FDA change-control expectations require that each such change be re-assessed for revalidation, data exclusion, or protocol action [3,5]. A RIA dated only to study start is a snapshot of a trial that no longer exists by month six.
MYTH 4 “Only trials with a ‘real’ medical device need a governance function (a PMO).”
FACT If a phone alone can open four regulatory regimes and generate continuous RIA obligations, then every study using a connected device needs continuous governance — not just the ones with an obvious investigational device. A one-time QARA sign-off cannot track firmware versions, AI model drift, country-by-country operator roles, and change control across a live program. That is structurally a program-management problem, which is why DHT programs increasingly run a QARA-aware PMO as a standing governance layer rather than a single assessment [1,8]. “Mobile-device-only” is not “governance-optional.” It is the case that needs governance the most, because nobody is watching the phone.
MYTH 5 “If it’s not in EUDAMED, the obligations aren’t ours.”
FACT Registration status does not create or excuse obligations — activity does. If you import a device into the EU for your trial, you can inherit economic-operator duties (importer, distributor, even labeller) under EU MDR Articles 13–16 the moment the device crosses the line, whether or not anyone has filed anything [3]. A sponsor once reused a CE-marked thermometer across three EU countries without updating the Instructions for Use (IFU) or assigning an Authorised Representative; the result was a three-month activation delay and a full re-submission [4]. The absence of a registration record is not the absence of liability. It is usually the first audit finding.

Why This Quietly Costs You
The danger of “it’s just a phone” is not that it’s wrong in an obvious way. It’s that it’s wrong invisibly. Nothing breaks at feasibility. Nothing breaks at first patient in. The gap between “works in theory” and “passes inspection” stays hidden until an inspector asks a deceptively simple question — “Can you walk me through this Digital Health Technology?” — and suddenly everyone remembers a meeting they meant to schedule.
By the time the device is in a participant’s hand, the regulatory roles are already yours [2]. The teams who win are not the ones with the flashiest devices; they are the ones who decided, early, that the boring questions — Who classified this? What’s its context of use? Who re-assesses it when it changes? Who owns that across the whole program? — deserved a real answer and a real owner.
The Bottom Line
“It’s just a phone” is a conclusion, not a starting point. The starting point is a Regulatory Impact Assessment that treats the device by what it does, repeats every time it changes, and lives inside a governance function that doesn’t care whether the study was labelled “device” or not. Get those three things right and the phone really can become boring — which, in clinical research, is the highest compliment a device can earn.
References
U.S. Food and Drug Administration. Digital Health Technologies for Remote Data Acquisition in Clinical Investigations. Guidance. 2023.
U.S. Food and Drug Administration. Framework for the Use of Digital Health Technologies in Drug and Biological Product Development. 2023.
European Parliament and Council. Regulation (EU) 2017/745 on Medical Devices (EU MDR). Official Journal of the European Union; 2017.
European Federation of Pharmaceutical Industries and Associations. Reflection paper on integrating medical devices into medicinal product clinical trials. Brussels: EFPIA; 2025.
IEC 62304:2006/AMD1:2015. Medical device software — Software life cycle processes. Geneva: IEC.
ISO 14971:2019. Medical devices — Application of risk management to medical devices. Geneva: ISO; 2019.
European Parliament and Council. Regulation (EU) 2024/1689 laying down harmonised rules on artificial intelligence (AI Act). 2024.
International Council for Harmonisation. ICH E6(R3): Good Clinical Practice. 2023.