From Compliance Burden to Competitive Edge: Why DHT-Ready Trials Win Faster Approvals
How quality, governance, and digital rigor became the shortest path to regulatory success

Introduction
The Great Misunderstanding About Compliance
Here is a contrarian claim for clinical operations professionals: the most compliant sponsors are now the fastest sponsors.
For years, the industry has treated compliance as a tax on innovation. It slows teams down. It adds cost. It fills binders that no one wants to read. Digital Health Technologies (DHTs)—wearables, sensors, eCOA apps, point-of-care devices, and algorithms—were supposed to change that by making trials faster, more patient-centric, and more efficient [1,2].
But as DHTs moved from pilot curiosities to primary evidence generators, regulators raised the bar. And something unexpected happened.
Organizations that invested early in DHT-ready governance did not slow down. They sped up. They received fewer questions. They avoided rework. They cleared reviews faster [1–3].
In today’s regulatory environment, compliance is no longer the opposite of speed—it is the enabler of it.
This article argues that sponsors who build DHT compliance into trial design—not onto it—can cut 3–6 months from development timelines. The data, the regulatory signals, and the operational evidence all point the same way.
1. The Old Model: Compliance as a Cost Centre
Traditional clinical compliance was largely reactive.
Validate systems because auditors expect it.
Write SOPs because QA requires them.
Answer regulator questions when they arise.
In this model, compliance happens late, adds friction, and is perceived as overhead. This mindset persisted into early digital trials, where sponsors treated DHTs as “just another data source” and assumed existing GCP frameworks would stretch to cover them [3,4].
They didn’t.
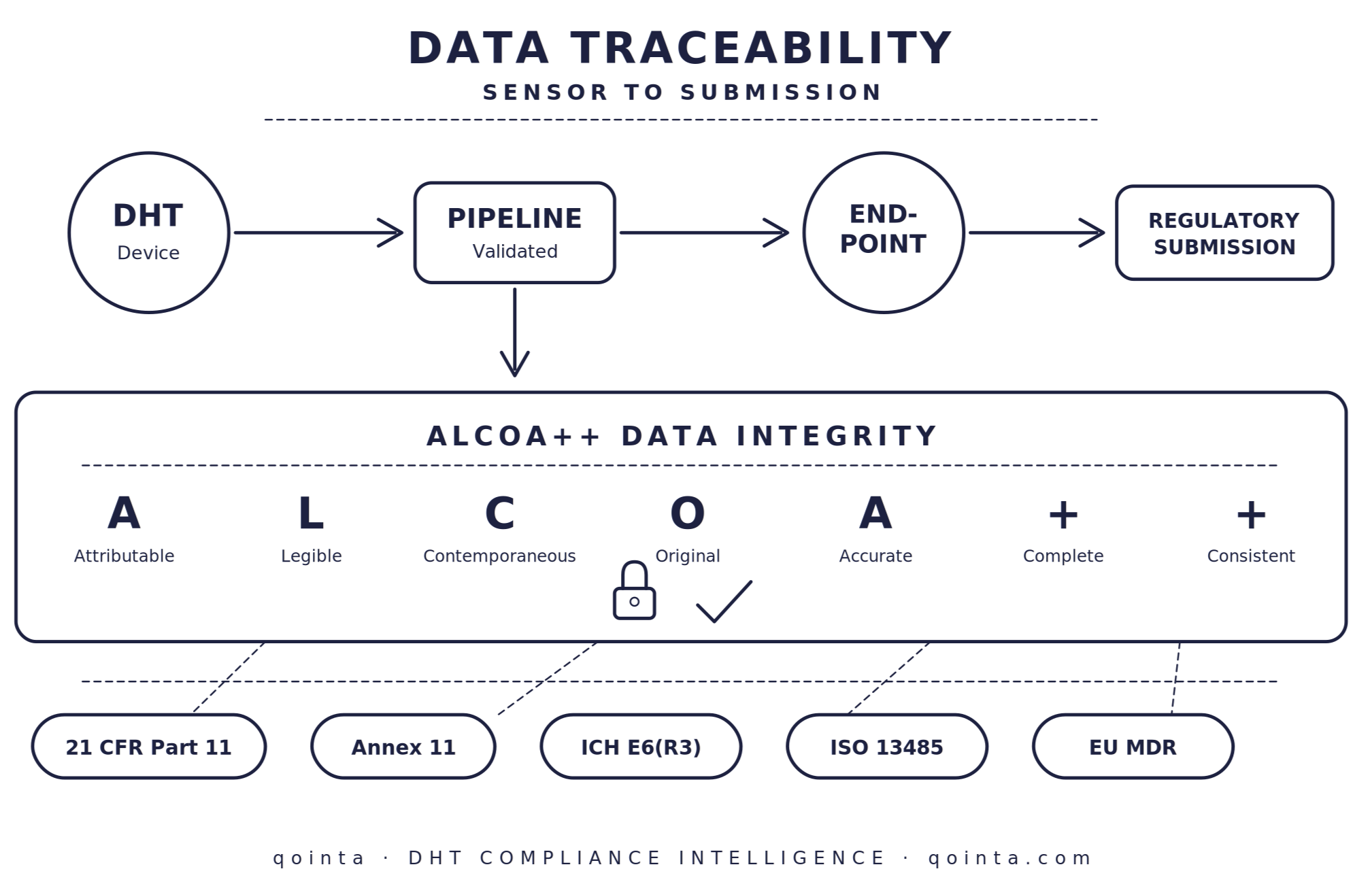
FDA guidance on Digital Health Technologies and the Framework for DHT Use now make explicit that devices, software, algorithms, and data pipelines must be governed as regulated instruments—not IT accessories [1,2]. The European Medical Device Regulation (EU MDR 2017/745) further tightened the net by redefining software, including clinical trial tools, under medical device law [5]. ICH E6(R3) extends the emphasis on technology oversight and data integrity under Good Clinical Practice [4].
The result: teams that delayed governance paid later—with protocol amendments, data exclusions, and extended review cycles.

2. The Shift: DHT Readiness as an Approval Accelerator
A new pattern is emerging across FDA, EMA, and MHRA interactions. Trials that demonstrate the following characteristics encounter fewer regulatory questions and faster alignment:
Fit-for-purpose DHT validation (ISO 14971, IEC 62366-1) [6,7]
Stable, scientifically defensible endpoint definitions
Controlled firmware, app, and algorithm change management
End-to-end data traceability enforcing ALCOA++ principles [3]
Clear role ownership across sponsors, CROs, vendors, and device manufacturers [5]
Why does this matter? Because regulators are no longer spending review cycles trying to understand whether the data can be trusted. That question has already been answered. DHT readiness removes uncertainty from the review process—and uncertainty is what slows approvals [1,2,8].
3. Why Regulators Move Faster When Digital Governance Is Strong
Regulatory review time is not driven by page count. It is driven by confidence.
When regulators see DHT-ready trials, they see measurement instruments that behave consistently over time, endpoints that are scientifically defensible, change control that prevents silent drift, and data lineage that can be reconstructed without speculation [1–3].
FDA’s increasing emphasis on fit-for-purpose validation, algorithm transparency, and data integrity reflects this shift [1,2,8]. The EMA’s landmark qualification of wearable-derived stride velocity as a primary endpoint in Duchenne muscular dystrophy studies demonstrated that robust validation frameworks accelerate—rather than hinder—regulatory acceptance of novel digital endpoints [9].
A regulator who trusts the system does not need to interrogate every output.
THE CONTRARIAN INSIGHT
The 3–6 months that sponsors typically save is not found by cutting corners. It is found by removing doubt. Every unanswered question about device validation, algorithm drift, or data lineage adds weeks to a review cycle. DHT-ready sponsors answer those questions before they are asked.
4. What “Good” Looks Like: The DHT-Ready Trial Architecture
Organizations that consistently outperform peers share a recognisable operating model.
4.1 A DHT-Ready Quality Management System
Their QMS extends beyond GCP to include device lifecycle governance (ISO 13485), software lifecycle controls (IEC 62304), risk management for digital endpoints (ISO 14971), human-factors validation (IEC 62366), and digital ALCOA++ enforcement across all systems [3,5–7]. This creates a single quality backbone for device, data, and clinical operations.
4.2 Endpoint-First Design
Digital endpoints are designed with explicit measurement rationale, predefined handling of missingness and drift, and locked algorithm versions for primary analyses [8,9]. This reduces post-hoc justification during review.
4.3 Controlled Change, Not Frozen Innovation
Firmware, app, and algorithm changes are assessed for endpoint impact, documented with scientific rationale, and implemented only when equivalence is demonstrated [1,2]. Regulators accept change—but not surprise.
4.4 Global Role Clarity
DHT-ready organizations map manufacturer, system producer, importer, and distributor roles, as well as data controller vs. processor responsibilities, before device deployment [5]. This prevents last-minute compliance gaps during global submissions.
5. The Economics of Being DHT-Ready
While few sponsors track it explicitly, the cost of not being DHT-ready is substantial.
| Cost of NOT Being DHT-Ready |
Returns of DHT Readiness |
|---|---|
| Extended review timelines due to data questions |
Fewer major review questions |
| Reanalysis caused by endpoint instability |
Reduced inspection findings |
| Protocol amendments late in development |
Faster alignment on endpoint acceptability |
| Lost credibility for future digital claims |
Greater regulator willingness to engage early |
| 3–6 months added to timelines |
3–6 months saved on timelines |
Compliance, in this context, becomes a return-on-investment strategy, not a cost centre [1–3].
6. Why This Advantage Will Compound
As digital endpoints become more common, regulators will benchmark expectations against the strongest submissions—not the weakest. This creates a compounding effect: DHT-ready sponsors set the bar, others are measured against it, and review tolerance narrows [1,2,8].
The FDA’s PDUFA VII commitments, the creation of the Digital Health Center of Excellence, and the expanding DHT Steering Committee all signal that digital governance expectations will only increase [1,10]. In Europe, the Clinical Trials Regulation (EU 536/2014) and the evolving EU AI Act further reinforce that algorithm transparency and data traceability are non-negotiable [5,11].
By 2027, digital governance will no longer be a differentiator. It will be table stakes.
The competitive edge belongs to those who build it now.
7. A Practical Readiness Check
If your next regulatory submission were tomorrow, could you answer these questions calmly and completely?
How was each digital endpoint generated?
Which device, firmware, and algorithm versions were used?
What changed mid-trial, and how did you assess endpoint impact?
Who was responsible for each component of the digital system?
How do you know data collected across multiple countries are equivalent?
Could you reconstruct every digital endpoint end-to-end for an inspector?
If any answer requires improvisation, the organisation is not DHT-ready.
Conclusion: Compliance Is the New Speed
The fastest approvals are no longer won by cutting corners. They are won by removing doubt.
DHT-ready trials succeed because they replace regulatory uncertainty with confidence—confidence in devices, algorithms, data, and decisions. In the modern clinical landscape, compliance is no longer the cost of innovation. It is the infrastructure that allows innovation to move faster [1–4].
The organisations that recognise this will not just meet regulatory expectations. They will outpace their competitors—quietly, consistently, and credibly.
The question is no longer whether compliance slows you down. The question is whether your competitors have already figured out that it speeds them up.
References
U.S. Food and Drug Administration. Digital Health Technologies for Remote Data Acquisition in Clinical Investigations: Guidance for Industry. Silver Spring (MD): FDA; 2023.
U.S. Food and Drug Administration. Framework for the Use of Digital Health Technologies in Drug and Biological Product Development. Silver Spring (MD): FDA; 2023.
U.S. Food and Drug Administration. Data Integrity and Compliance With Drug CGMP: Questions and Answers. Silver Spring (MD): FDA; 2018.
International Council for Harmonisation. ICH E6(R3) Good Clinical Practice (Draft Step 3). Geneva: ICH; 2023.
European Union. Regulation (EU) 2017/745 of the European Parliament and of the Council on Medical Devices (EU MDR). Official Journal of the European Union; 2017.
International Organization for Standardization. ISO 14971:2019 — Medical Devices: Application of Risk Management to Medical Devices. Geneva: ISO; 2019.
International Electrotechnical Commission. IEC 62366-1:2015+AMD1:2020 — Medical Devices: Application of Usability Engineering to Medical Devices. Geneva: IEC; 2020.
Kruizinga MD, Stuurman FE, Groeneveld GJ, et al. Development of Novel, Value-Based, Digital Endpoints for Clinical Trials: A Structured Approach Toward Fit-for-Purpose Validation. Pharmacol Rev. 2020;72(4):899–909.
European Medicines Agency. Qualification Opinion on Stride Velocity 95th Centile as a Secondary Endpoint in Duchenne Muscular Dystrophy. Amsterdam: EMA; 2019.
U.S. Food and Drug Administration. PDUFA VII Commitment Letter: Enhancing Use of Digital Health Technologies to Support Drug Development and Review. Silver Spring (MD): FDA; 2022.
European Parliament and Council. Regulation (EU) 2024/1689 Laying Down Harmonised Rules on Artificial Intelligence (EU AI Act). Official Journal of the European Union; 2024.