The May 2026 EUDAMED Deadline: What It Means for Digital Health Technologies in Clinical Trials
A readiness self-audit for sponsors, CROs, and manufacturers deploying wearables, sensors, and SaMD in EU clinical studies
Introduction: The Clock is Ticking

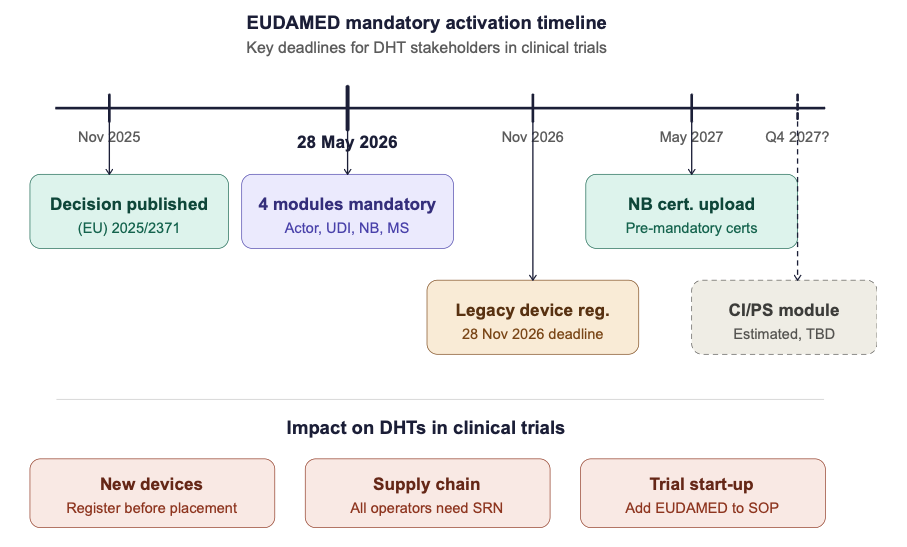
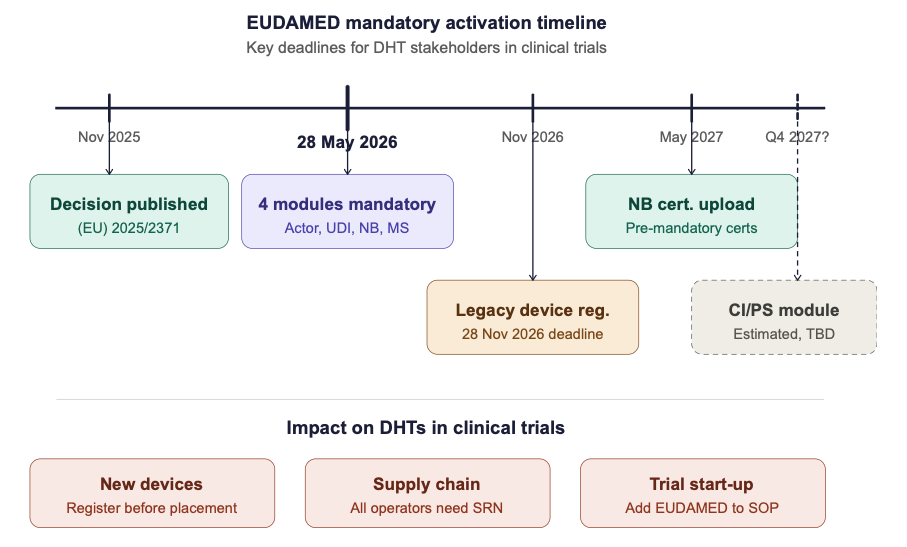
On 27 November 2025, the European Commission published Decision (EU) 2025/2371 in the Official Journal of the European Union, formally declaring four of the six EUDAMED modules fully functional [1]. That single publication triggered a six-month countdown. From 28 May 2026, the use of these modules becomes a legal requirement for every economic operator placing medical devices on the EU market [2].
For companies running clinical trials with digital health technologies (DHTs) — wearables, connected sensors, Software as a Medical Device (SaMD), and similar tools — this is not a distant regulatory milestone. It is an operational deadline with concrete consequences. If your DHT qualifies as a medical device under the EU Medical Device Regulation (MDR 2017/745), it must be registered in EUDAMED before market placement starting 28 May 2026 [3].
This article is structured as a readiness self-audit. Use it to evaluate where your organisation stands, identify gaps, and build a concrete action plan before the window closes.
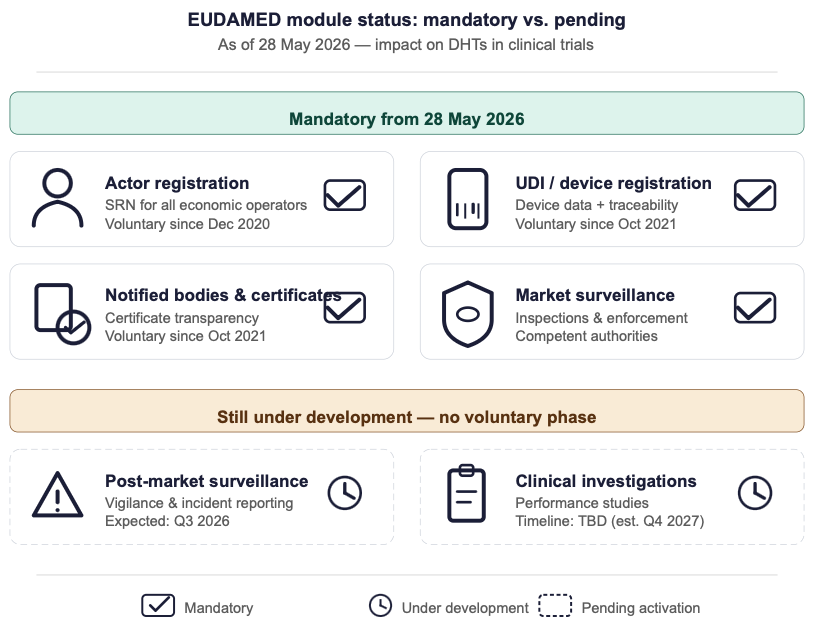
1. What Exactly Becomes Mandatory?
The European Commission confirmed that the following four EUDAMED modules now meet the functional requirements of Article 34(2) MDR [1]:
| Module |
What It Covers |
|---|---|
| Actor Registration |
Economic operators (manufacturers, authorised representatives, importers) must register and obtain a Single Registration Number (SRN). Without an SRN, no regulatory activity in EUDAMED can proceed [3]. |
| UDI/Device Registration |
Devices placed on the EU market must be registered with Unique Device Identification (UDI) data, supporting traceability and post-market monitoring [2]. |
| Notified Bodies & Certificates |
All certificates issued under MDR/IVDR must be uploaded. Refused applications become visible to all Notified Bodies [4]. |
| Market Surveillance |
Competent Authorities use this module to coordinate inspections, enforcement actions, and non-compliance reports across Member States [1]. |
Key distinction: Two modules remain under development — the Post-Market Surveillance and Vigilance (VGL) module and the Clinical Investigations/Performance Studies (CI/PS) module. These will be released only when they become mandatory, with no voluntary use phase before activation [5]. The Vigilance module is expected to be declared functional in Q3 2026, while the Clinical Investigations module is still under development [6]. Until then, clinical investigation applications and vigilance reporting must continue through national processes.
2. Why This Matters for DHTs in Clinical Trials
Digital health technologies occupy a uniquely complex position in this regulatory transition. Unlike traditional bench-top laboratory devices, DHTs in clinical trials often sit at the intersection of several regulatory frameworks:
Device classification under MDR Rule 11 (which often pushes software into Class IIa or higher) [7].
Clinical trial regulations under MDR Article 62 (clinical investigations of medical devices) [8].
Drug trial regulations when DHTs serve as non-investigational devices supporting pharmaceutical endpoints [9].
Data protection requirements under GDPR for patient-facing connected technologies [10].
The critical question every sponsor, CRO, and device manufacturer must answer: Does your DHT qualify as a medical device under MDR? If it does, EUDAMED registration becomes a prerequisite for market placement from 28 May 2026 [3]. This applies whether the device is the subject of the clinical investigation or is deployed as a non-investigational tool for endpoint capture, safety monitoring, or remote data acquisition.
Consider this scenario: A pharma sponsor deploys a CE-marked wearable sensor in a Phase III oncology trial to capture gait-related endpoints. The sensor manufacturer is a non-EU company with an EU Authorised Representative. Under the new EUDAMED obligations, the manufacturer must have completed Actor Registration and obtained an SRN. The device must be registered in the UDI/Device module. And the Authorised Representative must be registered as an economic operator. If any of these steps are incomplete by 28 May 2026, the device cannot legally be placed on the EU market — even if it carries a valid CE mark [2,3].
3. EUDAMED Readiness Self-Audit for DHT Stakeholders
Use the checklist below to assess your organisation’s preparedness. Each item maps directly to a mandatory EUDAMED obligation. Items marked as incomplete require immediate action.
Audit Area 1: Actor Registration
☐ Is your organisation registered in the EUDAMED Actor module?
☐ Have you obtained a Single Registration Number (SRN)?
☐ Is your EU Authorised Representative (if applicable) also registered with their own SRN?
☐ Are all economic operators in your supply chain (importers, distributors) registered?
☐ Is your Person Responsible for Regulatory Compliance (PRRC) information submitted? [3]
Why it matters: Without an SRN, no device registration can proceed. National Competent Authorities validate submissions, and a surge in registrations may create backlogs. Delayed SRN issuance will block device registrations and market access [3].
Audit Area 2: UDI/Device Registration
☐ Have you assigned UDI-DIs to all devices requiring EUDAMED registration?
☐ Is your UDI data accurate and complete (intended purpose, classification, conformity assessment route, applicable certificates)?
☐ Are new devices scheduled for EU market placement after 28 May 2026 prepared for registration before placement? [2]
☐ Are legacy devices (placed on market before 28 May 2026) scheduled for registration by 28 November 2026? [2]
☐ For large portfolios, have you evaluated Machine-to-Machine (M2M) solutions for bulk upload? [3]
Audit Area 3: Notified Body & Certificate Alignment
☐ Has your Notified Body confirmed their timeline for uploading certificates to EUDAMED?
☐ Are you aware that refused certificate applications will be visible to all Notified Bodies? [4]
☐ Do you maintain accessible copies of certificates for distributors or importers during the interim period? [4]
Timeline note: Notified Bodies must upload certificates issued prior to 28 May 2026 by 28 May 2027 [4]. Certificates issued after the mandatory date must be registered immediately upon issuance.
Audit Area 4: Trial-Specific Considerations
☐ Is your DHT classified as a medical device under MDR? If so, is the classification documented and defensible? [7]
☐ For non-investigational DHTs in drug trials: have you determined whether MDR Article 62 applies? [8]
☐ Have you mapped economic operator roles (manufacturer, Authorised Representative, importer, distributor) for every jurisdiction? [11]
☐ Are your vendor agreements updated to reflect EUDAMED registration obligations?
☐ Have you assessed the impact of EUDAMED registration on trial start-up timelines in EU Member States?
4. Key Deadlines at a Glance
| Deadline |
Obligation |
Who |
|---|---|---|
| 28 May 2026 |
Actor Registration + SRN required |
All economic operators |
| 28 May 2026 |
New devices: registered before market placement |
Manufacturers, ARs |
| 28 November 2026 |
Legacy devices: registered if still marketed |
Manufacturers, ARs |
| 28 May 2027 |
Pre-mandatory certificates uploaded |
Notified Bodies |
| TBD (est. Q4 2027) |
Clinical Investigations module mandatory |
Sponsors, investigators |
5. The SaMD Question: When Software Becomes a Registration Obligation
Software as a Medical Device deserves special attention. Under MDR Rule 11, software intended to provide diagnostic or therapeutic information is classified at minimum as Class IIa — unless the decision it supports could lead to serious deterioration or surgical intervention, in which case it escalates to Class IIb or III [7]. This classification has profound implications for EUDAMED compliance:
Conformity assessment via a Notified Body is required for Class IIa and above. The resulting certificate must appear in the EUDAMED NB/Certificates module [4].
CE marking prerequisites now include EUDAMED registration. A device cannot legally carry a CE mark for EU market placement without being registered from 28 May 2026 [2].
Software updates and version changes that affect intended purpose or risk classification may require updated EUDAMED entries, creating lifecycle management obligations beyond initial registration [12].
For sponsors deploying SaMD in clinical trials — whether for ePRO capture, clinical decision support, or real-time monitoring — the question is no longer whether the software needs to be registered, but whether your SaMD vendor has completed the registration process.
6. Immediate Action Plan: What to Do Now
Step 1: Confirm device classification. For every DHT used in your trials, determine whether it qualifies as a medical device under MDR. Document the rationale, especially for borderline cases involving general wellness devices repurposed for clinical endpoints [7].
Step 2: Verify Actor Registration. Contact your device manufacturers and confirm they hold a valid SRN. If they operate through an EU Authorised Representative, verify that entity is also registered [3].
Step 3: Audit the supply chain. Map every economic operator role — manufacturer, Authorised Representative, importer, distributor — across every EU jurisdiction where the trial operates. Each must be registered [11].
Step 4: Coordinate with Notified Bodies. Confirm certificate upload timelines and ensure pre-mandatory certificates are accessible during the transition period [4].
Step 5: Update trial documentation. Embed EUDAMED registration verification into trial start-up checklists, vendor qualification procedures, and Trial Master File requirements.
Step 6: Plan for the Clinical Investigations module. Although the CI/PS module is not yet active, its mandatory activation will follow without a voluntary use phase [5]. Begin aligning internal processes with the anticipated requirements now.
7. What Happens If You Are Not Ready?
The consequences of non-compliance are not theoretical:
Market access blocked: Devices without EUDAMED registration cannot be legally placed on the EU market from 28 May 2026 [2]. This directly affects trial supply and device deployment timelines.
Trial start-up delays: If a non-investigational device in a drug trial lacks registration, the sponsor may face ethics committee queries, regulatory authority holds, or site-level deployment blocks.
Audit exposure: Inspectors can cross-reference EUDAMED data during GCP inspections. A device used in a trial without proper registration may constitute a finding.
Double liability risk: Under Regulation (EU) 2024/1860, manufacturers must notify authorities about supply disruptions that could result in serious harm. Failure to update EUDAMED status for devices registered as ‘On the Market’ may trigger additional liability [3].
Conclusion: Registration Is the New Prerequisite
EUDAMED has been a work in progress for years. The voluntary use phase created an illusion of optionality. That illusion ends on 28 May 2026. For digital health technologies in clinical trials, the message is unambiguous: if your device meets the definition of a medical device under MDR, EUDAMED registration is no longer a future consideration. It is a present-tense obligation.
The sponsors and manufacturers who act now — auditing their registrations, mapping their supply chains, coordinating with Notified Bodies — will preserve their ability to deploy devices in EU trials without interruption. Those who wait risk discovering, at the worst possible moment, that their perfectly validated device cannot legally be placed on the market it was designed to serve.
The deadline is 28 May 2026. The audit starts today.
References
[1] European Commission. Commission Decision (EU) 2025/2371 of 26 November 2025 on the notice regarding the functionality of EUDAMED modules. Official Journal of the European Union. 2025.
[2] European Commission. Overview: EUDAMED, the European Database on Medical Devices. Public Health. Retrieved December 2025. Available from: https://health.ec.europa.eu/medical-devices-eudamed/overview_en
[3] European Commission. Q&A on practical aspects related to the gradual roll-out of EUDAMED pursuant to MDR and IVDR, as amended by Regulation (EU) 2024/1860. 2025.
[4] BSI Group. EUDAMED first modules published. Press release. December 2025.
[5] Osborne Clarke. EU triggers mandatory Eudamed use for diagnostics and medtech from May 2026. Legal analysis. December 2025.
[6] BSI Group. Updated EUDAMED roadmap: Vigilance module expected Q3 2026; Clinical Investigations module under development. December 2025.
[7] European Commission. Regulation (EU) 2017/745 on medical devices (MDR). Annex VIII, Rule 11. Brussels: EC; 2017.
[8] European Commission. MDCG 2021-6: Guidance on clinical investigation under MDR. Brussels: EC; 2021.
[9] EFPIA. Reflection paper on integrating medical devices into medicinal product clinical trials. Brussels: EFPIA; 2025.
[10] European Parliament. Regulation (EU) 2016/679 — General Data Protection Regulation. Brussels; 2016.
[11] European Commission. Regulation (EU) 2017/745. Articles 13–16: Economic operator obligations. Brussels: EC; 2017.
[12] Celegence. EUDAMED Mandatory by May 2026: Key Steps for Manufacturers. Regulatory analysis. December 2025.