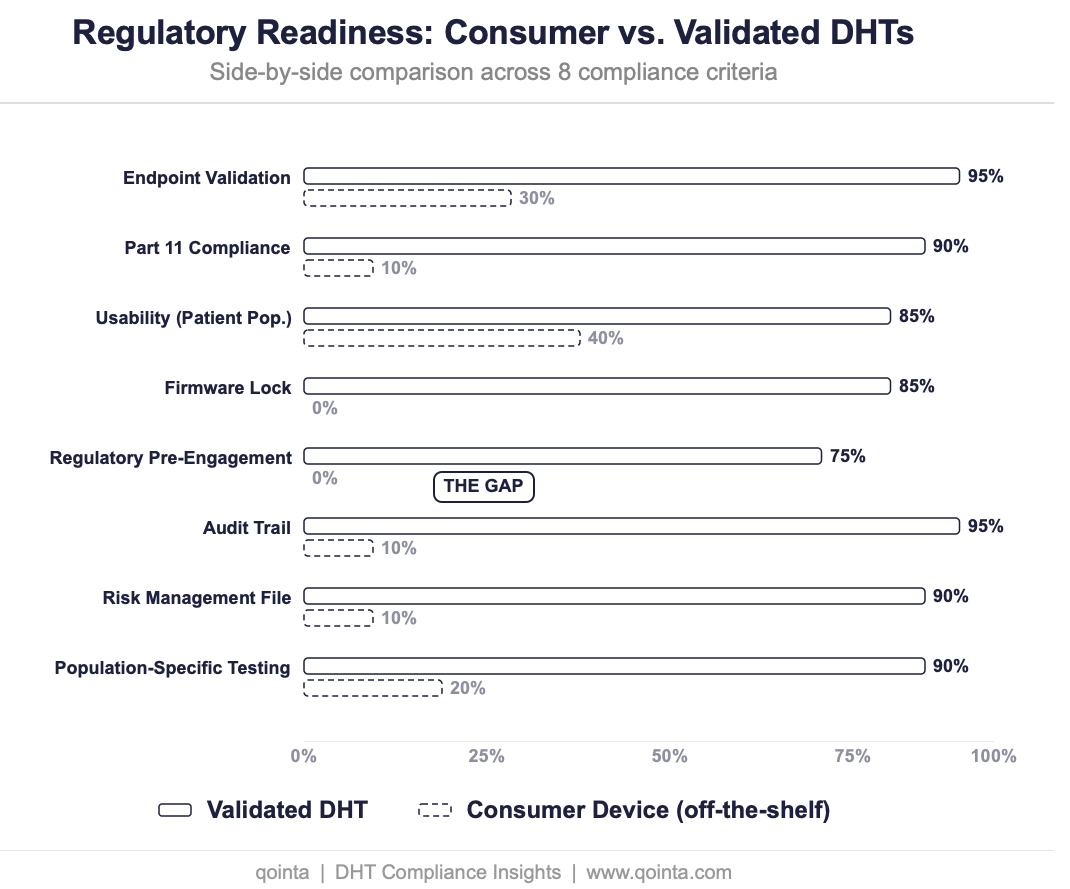
Are Consumer Devices (e.g. Fitbits, …) Compliant for Endpoint Data Collection?
FDA-cleared and trial-ready are not the same thing. Eight myths that could cost your next submission.
The Uncomfortable Question Nobody Wants to Ask

Consumer wearables are everywhere. Fitbits on wrists, Apple Watches pinging at board meetings, Garmin trackers logging morning runs. They’re familiar, affordable, and patients already own them. So when a clinical operations team asks, “Can’t we just use these for our trial?”—the question feels entirely reasonable.
The answer, however, is anything but simple. Consumer devices occupy a regulatory grey zone that catches sponsors off guard with remarkable consistency. The distinction between a device that is FDA-cleared for consumer wellness and a device that is fit-for-purpose in a regulated clinical trial is not a matter of semantics—it is a matter of whether your endpoint data will survive regulatory scrutiny [1,2].
This article dismantles eight persistent myths about using consumer wearables for endpoint data collection, replacing each with the regulatory and scientific reality that sponsors, CROs, and technology vendors must understand before the next protocol amendment.
The Regulatory Divide: Wellness vs. Medical Purpose
Consumer wearables are typically marketed under “general wellness” exemptions. A Fitbit tracking steps for personal fitness is not regulated as a medical device by FDA or EU MDR. However, the moment that same device is deployed in a clinical trial to generate endpoint data informing regulatory decisions, its regulatory identity transforms [1].
Under FDA guidance (2023), digital health technologies used in trials—even consumer-grade ones—must be fit-for-purpose validated [2]. The EU MDR 2017/745 can trigger Article 62 obligations when consumer devices are repurposed for medical endpoints [3]. And the MHRA requires UKCA marking considerations when device function extends beyond general wellness [4].
The bottom line: regulatory acceptance hinges on intended use, not consumer market status.
Challenges with Consumer Devices
Data Reliability Issues
Consumer wearables lack clinical-grade validation, and frequent firmware updates can alter algorithms mid-trial, compromising data consistency [5, 6].
Population-Specific Usability
Devices designed for healthy populations may underperform in patients with chronic conditions, such as Parkinson’s or COPD, leading to biased endpoint collection [7].
Data Integrity and Security
Consumer apps often lack built-in compliance with 21 CFR Part 11 (U.S. regulation for electronic records and signatures) or GDPR standards, exposing sponsors to audit risks [8].
Eight Myths That Could Derail Your Trial
| # | THE MYTH | THE FACT |
|---|---|---|
| 1 | "It's FDA-cleared, so we can use it in our trial." | FDA clearance for consumer use (e.g., 510(k) for general wellness) does not equate to fitness-for-purpose in a clinical trial. The FDA’s 2023 DHT guidance explicitly states that devices used for endpoint data must be validated for the specific clinical context, population, and endpoint—regardless of their market clearance status [2]. |
| 2 | "Consumer wearables capture the same data as medical-grade devices." | Consumer wearables are optimized for lifestyle utility, not clinical precision. Studies have shown that heart rate measurements from commercial fitness trackers can deviate significantly from reference-standard ECG monitors, particularly in patients with arrhythmias or darker skin tones [5]. Step counts and sleep staging algorithms vary between firmware versions, making longitudinal comparisons unreliable without strict version control. |
| 3 | "If it works for 400,000 people in the Apple Heart Study, it works for our trial." | The Apple Heart Study (2019) was an observational screening study—not a pivotal trial generating primary endpoints for regulatory labelling. Regulators acknowledged the study’s contributions while noting that findings were insufficient for product labelling claims [9]. Population-level screening and individual-level endpoint precision are fundamentally different regulatory asks. |
| 4 | "The device vendor guarantees accuracy, so validation is covered." | Vendor accuracy claims are typically derived from controlled bench testing with healthy volunteers. Clinical trials involve patients with comorbidities, polypharmacy, and variable adherence—conditions rarely replicated in vendor testing environments. Sponsors bear the regulatory burden of demonstrating that a device performs adequately in the specific study population [7]. |
| 5 | "Firmware updates are just improvements—they won’t affect our trial data." | Firmware and algorithm updates can silently alter how a device processes raw sensor data, changing heart rate smoothing windows, step-count thresholds, or sleep staging logic. An undocumented mid-study algorithm change can invalidate endpoint comparability across time points and introduce systematic bias that is nearly impossible to correct retrospectively [6]. |
| 6 | "Consumer devices are Part 11 compliant because they have cloud storage." | 21 CFR Part 11 compliance requires audit trails, electronic signatures, system validation, and access controls. Consumer cloud platforms were designed for personal convenience, not regulatory-grade record keeping. Without purpose-built integration with validated EDC or eSource systems, consumer device data lacks the provenance chain required for regulatory submissions [8]. |
| 7 | "Our patients prefer Fitbits, so regulatory acceptance will follow." | Patient preference is valuable for recruitment and retention, but regulators evaluate data quality, not user satisfaction. A device that patients love but that produces unvalidated endpoint data will not survive regulatory scrutiny. The bridge between patient preference and regulatory acceptance is rigorous fit-for-purpose validation [2,7]. |
| 8 | "CE marking means our device is compliant for European trials." | CE marking under EU MDR 2017/745 certifies conformity for the device’s declared intended purpose. When a consumer device is repurposed for clinical endpoint collection—a purpose outside its original declaration—it may trigger additional obligations under Article 62, including performance evaluation and potentially a new conformity assessment [3]. CE marking is a starting point, not a finish line. |
Evidence from the Field
Apple Heart Study (2019)
Enrolling over 400,000 participants via Apple Watches, this landmark study detected atrial fibrillation at unprecedented scale. Yet regulators were clear: the study’s observational screening design was not equivalent to endpoint validation, and findings did not support product labelling claims [9]. Scale is not a substitute for validation rigour.
Stride Velocity 95th Centile (EMA, 2019)
In Duchenne muscular dystrophy trials, wearable-derived stride velocity endpoints were accepted by the EMA as qualified secondary endpoints. The critical difference: these wearables underwent rigorous clinical validation specific to the trial population and endpoint, unlike off-the-shelf consumer alternatives [10].
COVID-19 Remote Monitoring Trials
During pandemic-era decentralized studies, sponsors deployed consumer oximeters and fitness trackers for safety monitoring. While operationally valuable, endpoints derived from these devices were not generally accepted by regulators without parallel validation against reference-standard instruments [6].
So What Should Sponsors Actually Do?
Consumer wearables are not automatically excluded from clinical endpoint collection. But sponsors must demonstrate fit-for-purpose compliance through a structured approach [2,11]:
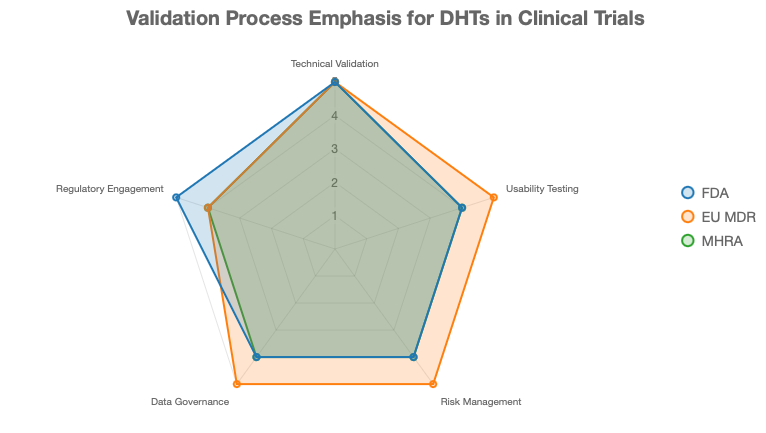
Technical validation: Benchmark device output against gold-standard clinical measures for the specific endpoint and population.
Usability testing: Confirm that your actual patient population can use the device reliably in uncontrolled settings.
Risk management: Align with ISO 14971 to identify and mitigate data integrity and patient safety risks [11].
Data governance: Ensure systems meet 21 CFR Part 11, HIPAA, and GDPR requirements with proper audit trails.
Regulatory engagement: Initiate early dialogue with FDA, EMA, or MHRA to justify device selection before the protocol is finalised.
The Verdict
Consumer devices like Fitbits offer real possibilities for cost-efficient, patient-friendly data capture in clinical trials. But they are rarely “compliant” in their off-the-shelf state. The gap between a wellness gadget and a regulatory-grade data collection instrument is bridged not by brand recognition or patient enthusiasm, but by validation, governance, and regulator engagement.
The next time someone on your team says “It’s FDA-cleared,” ask the follow-up question that matters: “But is it fit-for-purpose?”
References
FDA. General wellness: Policy for low risk devices. Guidance for industry. 2019.
FDA. Digital health technologies for remote data acquisition in clinical investigations. Guidance. 2023.
EFPIA. Reflection paper on integrating medical devices into medicinal product clinical trials. Brussels: EFPIA; 2025.
MHRA. Software and AI as a medical device change programme. London: MHRA; 2022.
Bent B, et al. The evaluation of commercial wearable devices for clinical trials. NPJ Digit Med. 2020;3:21.
Sehrawat O, Noseworthy PA, Siontis KC, et al. Data-driven and technology-enabled trial innovations toward decentralization. Mayo Clin Proc. 2023;98(9):1404–1421.
Aryal S, et al. Patient-centricity in digital measure development. NPJ Digit Med. 2024.
FDA. Part 11, electronic records; electronic signatures – Scope and application. Guidance. 2003.
Perez MV, et al. Large-scale assessment of a smartwatch to identify atrial fibrillation. N Engl J Med. 2019;381:1909–1917.
EMA. Qualification opinion: stride velocity 95th centile as secondary endpoint in Duchenne muscular dystrophy. EMA; 2019.
ISO 14971:2019. Medical devices – Application of risk management to medical devices. Geneva: ISO; 2019.